單細胞測序已成為生物醫學研究中最突出的技術之一。它在細胞水平上破譯轉錄組和表觀基因組變化的能力使研究人員獲得了有價值的新見解。因此,單細胞實驗的規模和復雜性增加了 100 多倍,涉及 100 多萬個細胞的實驗越來越普遍。
但是,必須在高度迭代的過程中對結果數據進行分析。至關重要的是,快速算法用于這些迭代步驟,以實現快速周轉時間。
為了使用 Python 進行更一致的單細胞分析,scverse致力于構建一個完整的生態系統,以幫助研究人員進行分析。該生態系統的核心是一種數據結構,它可以在整個數據處理管道中維護各種轉換的注釋,從而實現單細胞分析。
AnnData 是一個 Python 包,用于處理內存和磁盤上的注釋數據矩陣,是 Scanpy 庫,是 scverse 生態系統中的主要單細胞分析套件。Scanpy 構建在 PyData 生態系統中常見的其他庫之上,如 NumPy 、 SciPy 、 Numba 和 Scikit-learn,用于幾乎所有典型的分析步驟。
然而, Scanpy 算法大多是基于 CPU 的,并且在較大的實驗中速度顯著減慢。單細胞分析過程的高度迭代性質只會加劇這個問題。
GPU 用于單細胞分析
RAPIDS 可用于 GPU 進行下游單細胞 RNA 測序(scRNA-seq)分析的一般可行性,這一可行性已在 《使用 GPU 加速單細胞基因組分析》 中得到證實。此外,該工作還產生了 rapids-single-cell-examples GitHub repo,其中包含一系列由 RAPIDS 和 NVIDIA Parabricks 構建的示例。RAPIDS 是一個 GPU 的開源庫套件,用于 Python 的加速數據科學,而 Parabricks 是一套免費的 GPU 加速的、基于深度學習的行業標準基因組分析。
雖然這些示例筆記本在 GPU 上展示了一些典型的單細胞 RNA 工作流程,但它們從未用于日常使用,也從未被用作像 Scanpy 這樣的庫的 GPU – 加速替代品。
從以前的工作中汲取靈感,一個新興的圖書館叫 rapids-singlecell,是一種用于 scRNA 分析的 GPU 加速工具。該工具旨在成為與 scverse 生態系統兼容的每日可驅動單細胞分析套件,它使用 RAPIDS 和 CuPy 以提供 GPU 加速的函數,這些函數幾乎是 Scanpy 中相應函數的替代品。更多信息可以參考 rapids-single-cell-examples。
一般來說,用戶可以期望通過使用 RAPIDS,將性能提高 10 到 20 倍。想要了解更多信息,請訪問 使用 RAPIDS 加速單細胞基因組分析。
使用 RAPIDS 進行更快的單細胞分析
RAPIDS-singlecell 遵循與 scverse Python 庫類似的可用性模型。它也是用 Python 編寫的,但將許多性能關鍵部分放在 GPU 上,隱藏了通常與編寫 CUDA 應用程序(通常用于為 NVIDIA GPU 編寫加速算法的語言)相關的所有復雜性。
RAPIDS-singlecell 由五類組成,將在以下章節中進行描述。每個類別都加速了典型的單細胞分析工作流的不同部分。
想了解更多信息,包括 RAPIDS-singlecell 提供的各種 API,請訪問 rapids-singlecell 文檔。
cunData
AnData ,或注釋數據對象,是一種廣泛使用的數據結構,用于處理單細胞 RNA 測序數據。相反, cunData 是 GPU 的 AnData 對象的最小化和輕量級版本,它取代了用于預處理的 scverse 標準(圖 1 )。 cunData 不是將計數矩陣. X 存儲在 CPU 上,而是將其作為 CuPy 稀疏矩陣存儲在 GPU 上。這使得對計數矩陣執行計算更快、更高效。它還包含細胞(. obs 屬性)和基因(. var 屬性)的注釋數據幀,用于存儲細胞類型和基因名稱等附加信息。
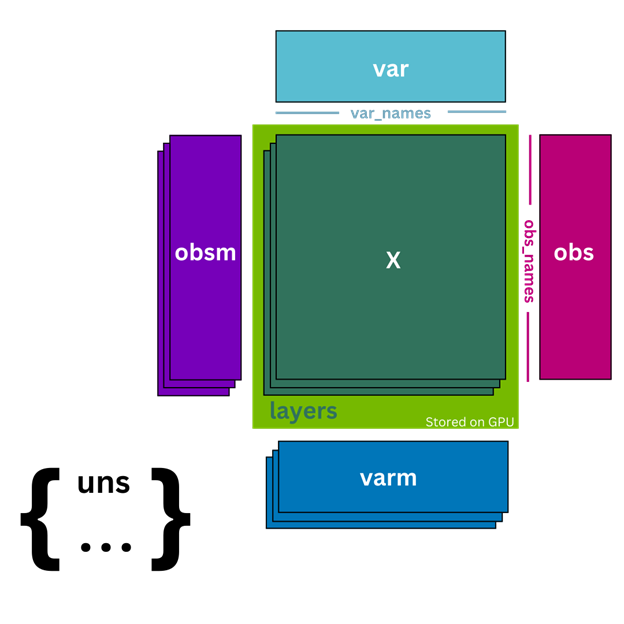
cunData 還包括其他功能,例如能夠將不同版本的計數矩陣(如原始整數計數)存儲在. Layers 中。與將. Layers 存儲在主機( CPU )內存中的 AnData 不同, cunData 還在 GPU 上存儲. Layers ,從而減少了將數據從主機復制到 GPU 內存的需要,并實現了加速計算。
cunData 支持. uns 屬性中的非結構化注釋,以及. obsm 和. vrm 屬性中的細胞和基因的多維注釋,這些注釋存儲在主機內存中。這些注釋使用戶能夠包括關于其數據的附加信息,例如空間坐標或主成分分析( PCA )嵌入。
類似地, cunData 支持像 AnnData 一樣的切片。但是,與視圖相反,這些切片始終是原始數據的完整副本。總體而言,與更具特征豐富的 CPU 結合的 AnnData 對象相比, cunData 能夠更快地預處理 scRNA-seq 數據。
下面的 Python 片段演示了 AnnData 對象(用于處理單細胞 RNA 測序數據的標準數據結構)到 cunData 對象的轉換。
import scanpy as sc
import rapids_singlecell as rsc
adata = sc.read("PATH TO DATASET")
cudata = rsc.cunnData.cunnData(adata=adata)
預處理
預處理函數存儲在cunnData_funcs,為 Scanpy 預處理功能提供了加速的替代方案。這些函數在cunData對象,并使用 RAPIDS cuML 和 CuPy 來顯著加速基于 Scikit-learn 、 NumPy 和 SciPy 的 Scanpy 函數。
過濾細胞和基因可以用filter_cells和filter_genes功能。質量控制由calculate_qc_metrics作用
# Basic QC rapids-singlecell rsc.pp.flag_gene_family(cudata,gene_family_name="MT", gene_family_prefix="mt-") rsc.pp.calculate_qc_metrics(cudata,qc_vars=["MT"]) cudata = cudata[cudata.obs["n_genes_by_counts"] > 500] cudata = cudata[cudata.obs["pct_counts_MT"] < 20] rsc.pp.filter_genes(cudata,min_count=3)
為了規范化您的數據, cunData _ funcs 提供了 GPU 替代normalize_total, log1p,以及最近推出的normalize_pearson_residualsScanpy 的函數。注釋highly variable genes對于 Scanpy 支持的所有口味(包括seurat,cellranger,seurat_v3,pearson_residuals)以及poisson_gene_selection,它改編自 scvi 工具。
# log normalization and highly variable gene selection cudata.layers["counts"] = cudata.X.copy() rsc.pp.normalize_total(cudata,target_sum=1e4) rsc.pp.log1p(cudata) rsc.pp.highly_variable_genes(cudata,n_top_genes=5000,flavor="seurat_v3",layer = "counts") cudata = cudata[:,cudata.var["highly_variable"]==True]
這個regress_out用 cuML 線性回歸估計器加速了用于去除不必要的變異源的函數。它還支持多目標回歸,這是在版本 22 . 12 的 cuML 中引入的,同時與以前的版本保持向后兼容。
cuML 包裝的主成分分析(PCA)、截斷奇異值分解(Truncated SVD)和增量主成分分析(Incremental PCA)為您提供與 Scanpy 提供的 PCA 函數 相同的選項。在 cunnData_funcs 中的 PCA 版本中,您可以選擇要用于分析的圖層,這是 Scanpy 目前不支持的附加功能。
# Regression, scaling and PCA rsc.pp.regress_out(cudata,keys=["total_counts", "pct_counts_MT"]) rsc.pp.scale(cudata,max_value=10) rsc.pp.pca(cudata, n_comps = 100) sc.pl.pca_variance_ratio(cudata, log=True,n_pcs=100)
cunndata_funcs可以將預處理加速 10 到 20 倍(表 1-3 )。經過預處理后, cunData 對象被轉換為 AnnData 對象。
adata_preprocessed = cudata.to_AnnData()
工具
scanpy_gpu提供了在 AnData 對象上工作的函數,目的是提供加速的函數。在 Scanpy 和 RAPIDS -singlecell 之間保持盡可能接近的語法,元數據也被寫入.uns屬性該屬性對于存儲經過訓練的參數(例如在 PCA 計算期間計算的方差比)非常有用。scanpy_gpu為 AnnData 對象提供了一個 PCA 函數,相當于cunnData_funcs.
Scanpy 已經支持使用 cuML 計算 GPU 上的 UMAP 和最近鄰居。scanpy_gpu通過添加更多算法,如基于加速圖的算法,擴展了對 Scanpy GPU 的支持clustering使用Leiden和Louvain來自 cuGraph ,以及Force Atlas 2用于直觀地布置圖形數據的算法。scanpy_gpu也使用PCA和kernel density estimation( KDE )來自 cuML 和diffusion maps以類似于 Scanpy 使用 SciPy 和 NumPy 進行科學計算的方式,使用 CuPy 庫進行計算。
對于批量校正,scanpy_gpu提供的 GPU 端口Harmony Integration,可以調用harmony_gpu。PyMDE(最小失真嵌入),一種能夠嵌入單細胞數據,同時以概率方式聯合學習圖和低維表示的函數,也可以在scvi-tools中使用。
RAPIDS – 單細胞置換性質的接近下降可以使用 Scanpy 進行可視化,使用起來很直觀,Scanpy 用于繪圖可以直接在 scverse 框架內進行。
解耦器
這個decoupler工具使用統一的框架實現了幾種不同的統計方法,重點關注生物活動(如細胞、分子和生理過程,例如基因集和轉錄因子活性)。decoupler_gpu重新實現并加速了加權和 (run_wsum) 以及多元線性模型 (run_mlm) 方法。RAPIDS 中的 GPU 端口-singlecell 使用與解耦器相同的網絡/模型。表 1 顯示 wsum 的性能提高了 37 倍。
Squidpy 開發
RAPIDS – 單細胞正在不斷擴展,為 scverse 生態系統提供新的加速功能。庫中添加了全面的測試,以確保代碼的正確性和可靠性。 Squidpy 能夠對空間分子數據進行詳細分析和可視化。它有助于理解復雜的細胞相互作用和空間模式,極大地促進了 scverse 生態系統的擴展。
RAPIDS 加速了一些功能,空間自相關與莫蘭的 I 和 Geary 的 C 承諾性能提升高達 100 倍。配體受體 (ligrec) 在 Squidpy 中的交互分析也得到了優化和加速,性能提升超過 10 倍。
基準
我們的基準測試結果表明,將 GPU 加速與 RAPIDS – 單細胞包和解耦器功能一起使用,可以顯著提高 scRNA-seq 分析的性能。
例如,運行示例 rapids-singlecell notebook,在具有兩個 AMD Epyc Milan 7543500GB 內存和一個NVIDIA A100 80GB GPU 的環境下,使用 RAPIDS-singlecell 包僅需 51 秒即可完成,而傳統的掃描 CPU 工作流程僅需 1106 秒。
類似地,解耦器功能也顯示出顯著的速度改進,與 CPU 上的 83 秒相比, GPU 上的 mlm 功能僅運行了 12 秒wsum方法在 GPU 上只需 26 秒,而在 CPU 上只需要 16 分 10 秒。
總之,這些結果證明了 GPU 加速使 scRNA-seq 分析更快、更有效的潛力。表 1 總結了這些基準結果。
| 作用 | CPU | GPU | 加速 |
| 整個筆記本電腦(不包括 PR 功能) | 1106 秒( 18 . 5 分鐘) | 51 秒 | 21 倍 |
| 預處理 | 74 秒 | 8 秒 | 9 倍 |
| HVG (Seurat v3) | 27 秒 | 1 . 6 秒 | 16 倍 |
| Regress out | 35 秒 | 0 . 7 秒 | 50 倍 |
| scale | 3 . 2 秒 | 0 . 4 秒 | 8 倍 |
| PCA | 417 秒 | 18 秒 | 23 倍 |
| Neighbors | 22 秒 | 5 . 1 秒 | 4 . 3 倍 |
| UMAP | 36 秒 | 0 . 4 秒 | 90 倍 |
| TSNE | 133 秒 | 2 . 4 秒 | 55 倍 |
| Louvain | 17 秒 | 0 . 6 秒 | 28 倍 |
| Leiden | 14 秒 | 0 . 2 秒 | 70 倍 |
| 邏輯回歸 | 58 秒 | 3 . 7 秒 | 15 倍 |
| 繪圖( FA2 ) | 256 秒 | 0 . 3 秒 | 850 倍 |
| run_mlm ( DoRothEA ) | 83 秒 | 12 秒 | 7 倍 |
| Run_wsum (程序) | 970 秒( 16 分鐘) | 26 秒 | 37 倍 |
除了之前的基準測試結果之外,運行一個示例rapids-singlecell 筆記本,當使用 RAPIDS-singlecell 時,服務器節點上的 500K 個 cell 大約只需要 2 分鐘。而在 CPU 上進行同樣的分析則需要 41 分鐘。
此外,使用pearson_residuals對于高度可變的基因選擇和標準化,也可以使用 GPU 加速,從而在 scRNA-seq 分析中提供額外的速度改進。這些基準結果匯總在表 2 中。
RAPIDS-singlecell 不僅能夠在高端服務器節點上加速單小區數據分析,而且能夠在消費級硬件上加速singlecell 數據分析。在 AMD 5950x CPU 、 64GB 內存和 NVIDIA RTX 3090 GPU 的臺式機系統上,使用 RAPIDS-singlecell ,端到端運行具有 50000 個電池的同一筆記本電腦大約需要 5 分鐘。盡管系統使用RAPIDS Memory Manager (RMM)和統一內存來超額訂閱 GPU 內存,但與 CPU 服務器相比,它的速度仍然顯著提高。這些基準結果匯總在表 2 中。
| 作用 | CPU | GPU ( A100 ) | GPU ( 3090 ) | 加速 |
| 整個筆記本(不包括公關功能) | 2460 秒( 41 分鐘) | 110 秒 | 290 秒 | 22 倍 |
| 預處理 | 305 秒 | 28 秒 | 169 秒 | 10 倍 |
| HVG (Seurat v3) | 48 秒 | 1 . 5 秒 | 13 秒 | 32 倍 |
| Regress out | 104 秒 | 5 . 1 秒 | 16 秒 | 20 倍 |
| scale | 8 . 4 秒 | 1 . 3 秒 | 5 秒 | 6 . 4 倍 |
| PCA | 86 秒 | 3 . 7 秒 | 35 秒 | 23 倍 |
| Neighbors | 74 秒 | 17 . 1 秒 | 18 . 3 秒 | 4 . 3 倍 |
| UMAP | 281 秒( 4 . 6 分鐘) | 6 . 7 秒 | 7 . 6 秒 | 60 倍 |
| TSNE | 786 秒( 13 分鐘) | 10 秒 | 12 . 9 秒 | 105 倍 |
| Louvain | 283 秒( 4 . 5 分鐘) | 4 . 5 秒 | 5 . 7 秒 | 62 倍 |
| Leiden | 282 秒( 4 . 5 分鐘) | 0 . 6 秒 | 0 . 9 秒 | 470 倍 |
| 邏輯回歸 | 452 秒( 7 . 5 分鐘) | 33 秒 | 63 秒 | 13 倍 |
| 擴散貼圖 | 30 秒 | 0 . 75 秒 | 1 . 3 秒 | 40 倍 |
| 重型車輛(公共車輛) | 104 秒 | 2 . 1 秒 | 15 . 6 秒 | 50 倍 |
| 規格化( PR ) | 22 秒 | 0 . 3 秒 | 1 秒 | 73 倍 |
當使用 RAPIDS -singlecell 時,在桌面系統上端到端運行具有約 90K 個單元格的相同示例筆記本(表 1 )僅需 48 秒。相比之下,傳統的掃描 CPU 工作流程需要 774 秒。加速解耦器功能還顯示出在消費級硬件上的顯著速度改進。表 3 總結了這些基準結果。
| 作用 | CPU | GPU | 加速 |
| 整個筆記本電腦(不包括解耦器功能) | 774 秒( 13 分鐘) | 48 秒 | 16 倍 |
| 預處理 | 114 秒 | 6 秒 | 19 倍 |
| Regress out | 62 秒 | 1 . 6 秒 | 39 倍 |
| 主成分分析 | 42 秒 | 0 . 7 秒 | 60 倍 |
| HVG (Seurat v3) | 2 . 7 秒 | 0 . 4 秒 | 6 . 7 倍 |
| PCA | 175 秒 | 21 . 7 秒 | 8 倍 |
| Neighbors | 14 . 9 秒 | 4 . 6 秒 | 3 . 2 倍 |
| UMAP | 31 秒 | 0 . 3 秒 | 103 倍 |
| TSNE | 95 秒 | 1 . 4 秒 | 68 倍 |
| Louvain | 9 . 3 秒 | 0 . 5 秒 | 18 倍 |
| Leiden | 13 . 2 秒 | 0 . 1 秒 | 130 倍 |
| 邏輯回歸 | 76 秒 | 3 . 75 秒 | 20 倍 |
| 繪圖( FA2 ) | 191 秒 | 0 . 23 秒 | 830 倍 |
| run_mlm ( DoRothEA ) | 55 秒 | 12 秒 | 4 . 5 倍 |
| Run_wsum (程序) | 690 秒( 11 . 5 分鐘) | 28 秒 | 26 倍 |
安裝
安裝 RAPIDS – 單電池有多種方法。最簡單的方法是使用 GitHub 存儲庫中提供的一個 yaml 文件。這些設置了整個環境,包括運行示例筆記本所需的一切。
conda create -f conda/rsc_rapids_23.02.yml
您還可以從 PyPI 將 RAPIDS -singlecell 安裝到 Conda 環境中,并從 Conda 安裝 RAPIDS 。默認安裝程序不包括 RAPIDS 或 CuPy 。 Scanpy 也被排除在外,因為它在技術上是不必要的。
pip install rapids-singlecell
最后,您可以使用 RAPIDS 中的新實驗 PyPI 包從 PyPI 安裝整個庫,包括 RAPDIS 依賴項。然而,這種安裝方法需要用戶正確設置 CUDA ,以便可以通過 RAPIDS 和 CuPy 找到它。
要執行此操作,可以使用以下命令:
pip install 'rapids-singlecell[rapids]’ --extra-index-url=https://pypi.nvidia.com
結論
帶 RAPIDS-singlecell 庫·,可以在比 CPU 僅計算其 UMAP 嵌入所花費的時間更短的時間內運行 500K 個小區的完整分析。因此,它能夠在單細胞數據分析階段實現更快的迭代過程。
RAPIDS-singlecell 還使生物信息學家能夠與醫生或生物學家實時分析數據,從而更好地協作和解釋數據。根據我們的經驗,即使在消費類 3090 系列顯卡上,也可以在沒有任何問題的情況下分析 200K 單元。更好的是, RMM 使 GPU 存儲器能夠被超額訂閱并溢出到主存儲器,從而使規模遠遠超過 500K 個單元。
使用數據中心類 NVIDIA A100 80 GB GPU ,您可以分析包含多達 2 個的矩陣31-1 (約 21 . 5 億)個非零計數。(請注意,這是用于稀疏矩陣計算的基于 CuPy 32 位整數的索引的當前限制。)這種強大的功能使用戶能夠分析超過 100 萬個單元格的數據集。
20 倍以上加速了 RAPIDS-singlecell 提供的功能使研究人員能夠更加專注于分析和解釋他們的單細胞數據,而不是等待漫長的計算過程。本著 RAPIDS 的真正精神,這最終提高了生產力,并促進了對細胞生物學的新見解,這在以前是不可能的。
?