蛋白質序列對齊 (比較蛋白質序列的相似性) 是現代生物學和醫學的基礎。它通過重建進化關系 (技術上稱為 homology inference) 來照亮基因功能,為藥物開發提供信息。當科學家發現或設計一種新蛋白質時,他們可以將其與已知的蛋白質序列進行比對,以推斷其結構和功能。
這種同源性搜索可以揭示有前景的藥物點 (例如,通過比較病原體蛋白質與人類蛋白質) 或查明致病突變 (通過比較患者的蛋白質與健康蛋白質) 。然而,基因組和宏基因組數據的快速擴展現在給傳統的對齊工具帶來了壓力。
本文將探討蛋白質對齊方面的最新進展如何通過使用 GPU 優化的對齊,以前所未有的速度增強 AI 驅動的藥物發現、結構預測和蛋白質設計,從而加速蛋白質科學。
蛋白質序列對齊擴展科學見解
序列對齊可能聽起來技術含量很高,但其重要性顯而易見:科學家可以比較蛋白質序列以找到相似性。相似序列通常意味著相似的功能或結構特征。這是同源性推理的基礎:如果蛋白質 A 與蛋白質 B 相似,則它們可能具有生物學作用。
蛋白質序列對齊通過識別保守區域、預測蛋白質功能和檢測可能導致疾病的不太可能的突變,對功能注釋、進化研究、疾病研究和藥物研發至關重要。在序列對齊中編碼的進化信息還可以指導藥物點的選擇和優化。

人類蛋白質組表現出極大的復雜性。典型的乳腺細胞包含大約 10 billion 個蛋白質分子,每個細胞的豐度動態范圍為 106。像 plasma 這樣的體液在豐富程度最高和最少的蛋白質之間表現出 1010 倍的差異。這種復雜性給全面的蛋白質組分析帶來了重大挑戰。
繪制蛋白質相互作用圖更加令人望而生畏。雖然人類蛋白質組中存在近 200 million 種可能的成對相互作用,但只有約 53,000 種經過實驗證實,這使得這種映射類似于在大海撈針。
從頭設計蛋白質是這些挑戰中計算復雜度最高的,涉及探索天文搜索空間 (N 長度蛋白質的 20N 序列) ,并解決折疊和功能優化中的 NP 難題。AI 和自動化的進步顯著提高了成功率 (在某些設計類別中,成功率從約 10% 提高到約 30 – 50%) ,但實驗驗證仍然需要大量資源,通常需要進行迭代測試和優化。雖然近期的突破加速了進展,但這些基本問題仍處于結構生物學的前沿。
從 BLAST 到 MMseqs2 的對齊演變
生物信息學家已經開發出越來越高效的算法來加速序列對齊。BLAST 在 20 世紀 90 年代徹底改變了搜索速度,但卻難以應對不斷增長的數據,因此在 2010 年代開發出了 DIAMOND 和 MMseqs2。
MMseqs2 的靈敏度優于 PSI-BLAST,運行速度提高了 400 多倍。在三次迭代的配置文件搜索中,MMseqs2 的速度比 PSI-BLAST 快 433 倍,同時還表現出更高的靈敏度。
MMseqs2 和 DIAMOND 現已廣泛應用于基因組注釋和藥物研發,取代了曾經需要數周計算才能完成的工作流中的 BLAST。然而,隨著數據量的持續爆炸,即使是基于 CPU 的最快工具也面臨著限制,這促使人們轉向 GPU 加速。盡管 AI 取得了進步,但蛋白質對齊仍然至關重要。AlphaFold2 等深度學習模型依靠多序列對齊 (MSA) 來預測蛋白質結構,從而證明快速且可擴展的序列搜索方法的持久重要性 (表 1) 。
| MMseqs2 用例 | 示例 | 參考資料 |
| 擴展和級聯 MSA 搜索以預測蛋白質結構 |
|
|
| 過濾冗余或同源序列 | ADOPT:識別內在無序蛋白質區域 | ADOPT:通過深度雙向Transformers預測內部蛋白質疾病 |
| 用于蛋白質相互作用分析的Clustering | 感知 – PPI | SENSE-PPI 以基因組尺度重建物種內部、之間和之間的相互作用組 |
在基于深度學習的工作流程中,更快的 MSA 具有巨大的潛在優勢。MSA 搜索的計算成本高昂,通常會占用推理和訓練時間。例如,AlphaFold2 和 OpenFold 的推理主要由 MSA 搜索進行 (約占總時間的 70 – 90%) ,與單個計算作業相比,將 MSA 對齊和折疊算法拆分成兩個作業可節省 AlphaFold 57% 的成本,而 OpenFold 可節省 51% 的成本。
MMseqs2-GPU 和下一次速度飛躍
MMseqs2-GPU 利用特定于 GPU 的新型加速在 CUDA 上解鎖多個序列對齊。開發 MMseqs2-GPU 的聯合研究團隊由首爾大學、Johannes Gutenberg University Mainz 和 NVIDIA 的研究人員領導。該團隊創建了一種針對 GPU 優化的新型“無縫”過濾算法,以取代基于 CPU k-mer 的預過濾器。簡單來說,GPU 版本并不像在 CPU 上掃描 BLAST 和 MMseqs2 那樣掃描短匹配子串 (k-mers) ,而是使用高度并行算法,直接在序列中對對齊進行評分,而沒有間隙 (允許不匹配,但沒有插入/ 刪除) 。
這種使用 CUDA 實施的方法經過專門定制,可避免 memory bottlenecks,同時使數千個 GPU 核心保持忙碌??。在此快速預過濾器找到有前景的匹配后,GPU 還可以針對這些命中執行更精確的 gapped alignment(使用優化的 Smith-Waterman 算法)。
為了全面了解這一進步,MMseqs2 GPU 技術報告中的主要性能比較包括以下內容:
- MMseqs2-GPU 可在 8 個 GPU 上實現高達 100 TCUPS (每秒萬億個單元更新) 的無縫過濾,比以前的加速方法高出 1 到 2 個數量級。
- 在單個 NVIDIA L40S GPU 上,與在 128 核 CPU 上運行以進行蛋白質序列搜索的 MMseqs2 k-mer 相比,MMseqs2-GPU 的速度提高了 20 倍,成本降低了 71 倍。
- 在 ColabFold 中,與使用 JackHMMER 的 AlphaFold2 相比,MMseqs2-GPU 在保持等效預測準確性的同時,將結構預測速度提高了 23 倍。
- 對于 Foldseek 中的蛋白質結構對齊,與基于 CPU 的版本相比,MMseqs2-GPU 可提供高達 27 倍的速度提升。
- 即使在 NVIDIA L4 等經濟高效的 GPU 上,MMseqs2-GPU 也能實現更快的同源搜索速度,在搜索 UniRef90 時,速度比 JackHMMER 快 10 倍。
序列對齊和蛋白質結構預測
作為 MMseqs2-GPU 在藥物研發工作流程中解決現實問題的示例,以下展示了序列對齊在 AI 驅動的目標結構預測中的作用。此處,MMSeqs2-GPU (MSA-Search) 和 OpenFold 模型均被打包為容器化的 NVIDIA NIM 微服務(圖 2)。
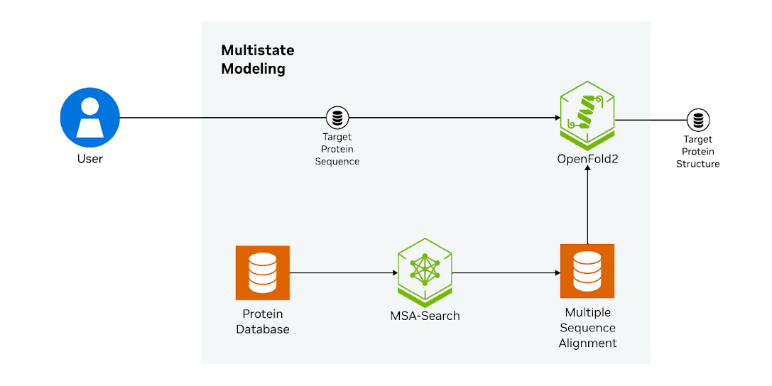
例如,MSA-Search NIM 和 OpenFold2 NIM 可以一起使用,對目標蛋白質的多個構象狀態進行建模。工作流程涉及以下步驟:
- 查詢 → 對齊:將 FASTA 序列發送到 MSA-Search NIM → 獲取 A3M MSA 和一個模板命中 HHR 文件。
- 選擇模板:選擇兩個 PDB IDs (打開和關閉) ,并提取其各自的 HHR 塊。
- 預測每種狀態:調用 OpenFold 2 NIM 兩次,每次傳遞相同的 A3M 但不同的 HHR 切片。
- 寫入結果:將返回的兩個 PDB 字符串另存為 hClpP_active.pdb 和 hClpP_inactive.pdb。
以下代碼以人類 hCLpP 蛋白質為例,說明了如何做到這一點:
"""Example: model two conformations of human ClpP (hClpP)"""import os, json, requests, pathlib# ------------------------------------------------------------------# Config# ------------------------------------------------------------------API_KEY = os.environ["NIM_API_KEY"]HEADERS = {"Authorization": f"Bearer {API_KEY}", "Content-Type": "application/json"}OF2_URL = "http://localhost:8000/biology/openfold/openfold2/predict-structure-from-msa-and-template"SEQ = """>hClpP_HUMANMARGKIIGELASKKKVEAMAAKLAEAG... (FASTA truncated for clarity)"""# ------------------------------------------------------------------# 1) Run GPU-MMseqs2 via MSA-Search NIM# ------------------------------------------------------------------msa_payload = { "sequence": SEQ, "databases": ["uniref90-2024_02", "pdb70"], "return_templates": True # tells the service to emit an HHR-formatted hit list}msa_resp = requests.post(MSA_URL, headers=HEADERS, data=json.dumps(msa_payload), timeout=900)msa_resp.raise_for_status()msa = msa_resp.json()a3m_alignment = msa["alignments"]["uniref90-2024_02"]["a3m"]["alignment"]# Helper: pick two PDB templates that represent distinct states.ACTIVE_PDB = "7DKF" # hClpP active/openINACTIVE_PDB = "7D7G" # hClpP inactive/closedhhr_all = msa["templates"]["pdb70"]["hhr"]["alignment"] # full HHR textdef slice_hhr_for(pdb_id: str, hhr_text: str) -> str: """Return an HHR minimal block for a single template PDB hit.""" keep = [] write = False for line in hhr_text.splitlines(): if line.startswith(">PDBID:") and pdb_id in line: write = True elif line.startswith(">PDBID:") and write: break if write: keep.append(line) return "\n".join(keep)hhr_active = slice_hhr_for(ACTIVE_PDB, hhr_all)hhr_inactive = slice_hhr_for(INACTIVE_PDB, hhr_all)# ------------------------------------------------------------------# 2) Predict *active* conformation with OpenFold 2# ------------------------------------------------------------------def run_openfold2(hhr_block: str, tag: str) -> pathlib.Path: payload = { "sequence": SEQ, "alignments": { "uniref90-2024_02": { "a3m": {"alignment": a3m_alignment, "format": "a3m"} } }, "templates": { "pdb70": { "hhr": {"alignment": hhr_block, "format": "hhr"} } }, "selected_models": [1] # run a single model for speed; omit for ensemble } r = requests.post(OF2_URL, headers=HEADERS, data=json.dumps(payload), timeout=1800) r.raise_for_status() pdb_text = r.json()["predictions"][0]["structure"] out = pathlib.Path(f"hClpP_{tag}.pdb") out.write_text(pdb_text) print(f"Wrote {out}") return outactive_pdb = run_openfold2(hhr_active, "active")inactive_pdb = run_openfold2(hhr_inactive, "inactive") |
GPU 加速生物信息學的未來方向
從 BLAST 到 MMseqs2-GPU,序列對齊方面的進步徹底改變了蛋白質科學,使人們能夠更快地了解功能、進化和藥物研發。
此工具已用于合成數據集生成(AI 模型開發生命周期的重要組成部分),以及用于測試時擴展和實時預測的加速推理。MMseqs2-GPU 已在業內得到廣泛采用,包括 Basecamp Research、VantAI 和 Iambic Therapeutics 等領先公司。
隨著 AI 驅動的模型將對齊集成到預測工作流程中,GPU 加速正在重新定義分子研究。AI、HPC 和生物信息學的融合有望實現更大的突破,加速醫學和生物技術的發現。詳細了解用于生成式蛋白質結合劑設計的 NVIDIA BioNeMo Blueprint。
試用 MMSeqs2-GPU 作為 NVIDIA MSA-Search NIM,并試用 OpenFold 作為 NVIDIA OpenFold2 NIM。
?